Full publication list available at
Google Scholar
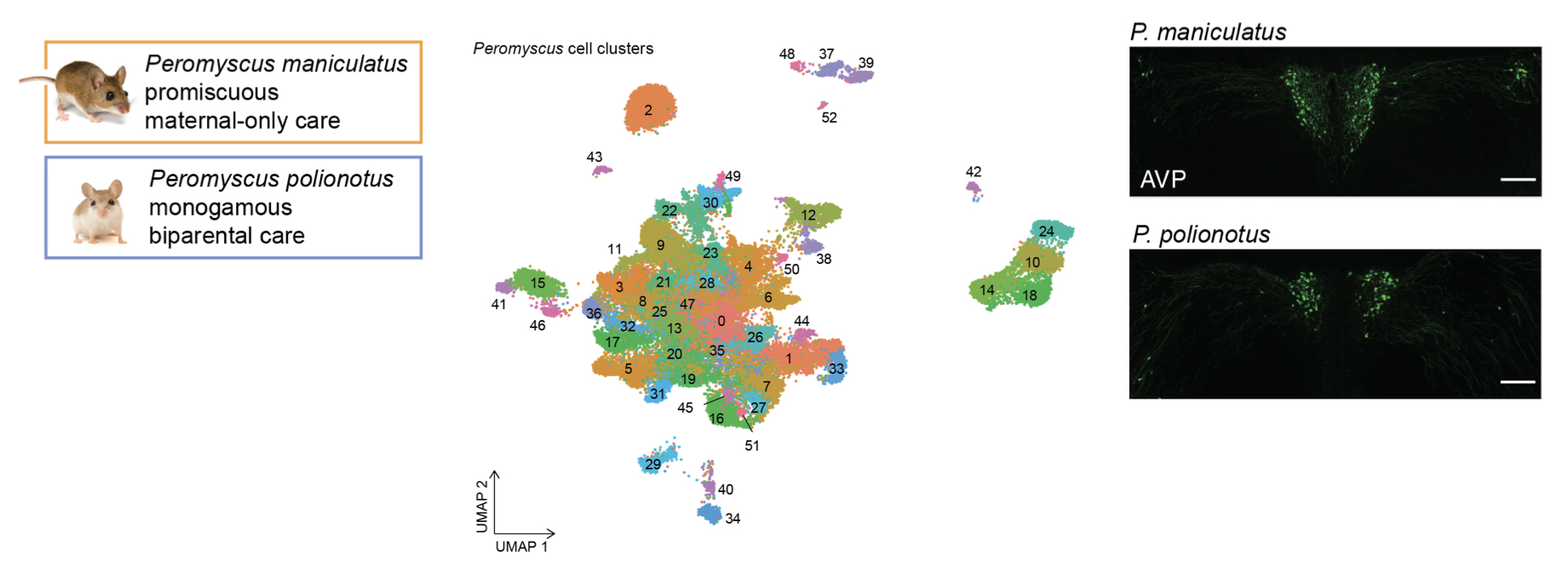
Chen J, Richardson PR, Kirby C, Eddy SR, and HE Hoekstra.
Cellular evolution of the hypothalamic preoptic area of behaviorally divergent deer mice. eLife. 2024; e103109.
Kautt A*,
Chen J*, Lewarch CL, Hu C, Turner K, Lassance JM, Baier F, Bedford NL, Bendesky A, and HE Hoekstra.
Evolution of gene expression across brain regions in behaviorally divergent deer mice and their hybrids. Molecular Ecology. 2024; e17270. *Contributed equally.
Chen J, Swofford R, Johnson J, Cummings BB, Rogel N, Lindblad-Toh K, Haerty W, di Palma F, and A Regev.
A quantitative model for characterizing the evolutionary history of mammalian gene expression.
Genome research. 2019; 29: 53-63.
Dixit A*, Parnas O*, Li B,
Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, Adamson B, Norman TM, Lander ES, Weissman JS, Friedman N, and A Regev.
Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens.
Cell. 2016; 167: 1853-1866.
Engreitz JM, Haines JE, Perez EM, Munson G,
Chen J, Kane M, McDonel PE, Guttman M, and ES Lander.
Local regulation of gene expression by lncRNA promoters, transcription and splicing.
Nature. 2016; 539: 452.
Chen J, Shishkin A, Zhu X, Kadri S, Hanna J, Regev A, and M Garber.
Evolutionary analysis across mammals reveals distinct classes of long noncoding RNAs.
Genome biology. 2016; 17: 19.